
Different omics have different levels of relevance
Proteins make the cell structure and function and cosmetics science knows it for years.
A gene gives the information that the translated protein can structure or work in the cell, but we now know that epigenetics mechanisms may interfere or inactivate gene. Genomics stays reasonably at the informational level. (fig.1)
The transcript (mRNA), reached by RNAseq at the quantitative shotgun level, tells us a protein may be translated but if and when remains an open question as post transcriptional mechanisms impact on the next steps.
Only proteomics tells us which protein is there, easily quantified now with nanoLC-MS/MS, a mass spectrometry technique.
Of course, no technique is able in an untargeted way to tell us if the protein as an enzyme is active and at what speed it produces metabolites, even if the post-translational modifications (PTMs) can be now reached by mass spectrometry (1). Also, no untargeted technique is able to know which protein interact with which other molecule.

Although transcriptomics, proteomics, and metabolomics all generally measure the products of gene expression, one should not expect correlation among them. Measuring the level of a transcript reflects the production rate of its protein product, but does not accurately predict the concentration pool or stability of the protein product. In fact, the correlation of mRNA abundances with their corresponding protein abundances, while reasonable for some core metabolic processes in some microbial systems, in general is poor or non-existent in most biological systems examined to date, suggesting proteomics data is likely more indicative of biological phenotype than mRNA. (4, 5)
Usually, to know the in vitro effects of a cosmetics or an ingredient, we compare cells or tissue samples which received the product to samples which received a placebo. It is the same with omics techniques where the relative quantitation of high number of impacted molecules will be studied. It is then crucial to use the omics which are the closest from the cell functioning, such as proteomics and metabolomics. (fig.2)

Integrated omics
To achieve a more complete understanding of metabolic activities, it is interesting to integrate -omics data. It is important to realize that while DNA and proteins are relatively stable, transcripts and metabolites often have very short half-lives; thus, there are dramatic temporal information differences in these omics measurements. By integrating these large-scale datasets, cellular metabolism can be examined at an upgraded information level. For complex samples such as microbiomes, this integrated omics information has potential to provide a detailed molecular view at a higher resolution (4). But in order to integrate multi-omics, the same kind of information is needed. Relative quantitation data of proteins can be integrated with mRNA relative quantitation data.
If not, for example at PHYLOGENE, having the information of major impacted taxons after a 16SrDNA profiling will focus the metaproteomics database queries on these taxons.
Associated bioinformatics.
For example, the current proteomics LC-MS/MS output data reach easily more than 5000 identified proteins. Including the microbial part of the skin, more than 10000 proteins can be reached easily in metaproteomics. The relative quantification of two conditions reveals tens to hundreds of under- or overexpressed proteins.
This requires heavy data processing pipelines (6) which will analyze all impacted proteins in correspondence with the metabolic pathways in which they are involved. It is so possible to understand the biological events induced by the disease.
Usually, a taxonomic analysis, a functional analysis by taxon (Homo sapiens, Fungi, Bacteria and Archae) and inter-functions correlations can be produced which give access to links between signaling/metabolic pathways, potential association of functions to particular taxons, potential interspecific relationships between microorganism and between microorganism and human (7). As a synthesis, the mechanism of action can be formulated (8).
Fully integrated substantiation studies in cosmetics
PHYLOGENE expertise is based on the knowledge of the using of omics techniques for 20 years.
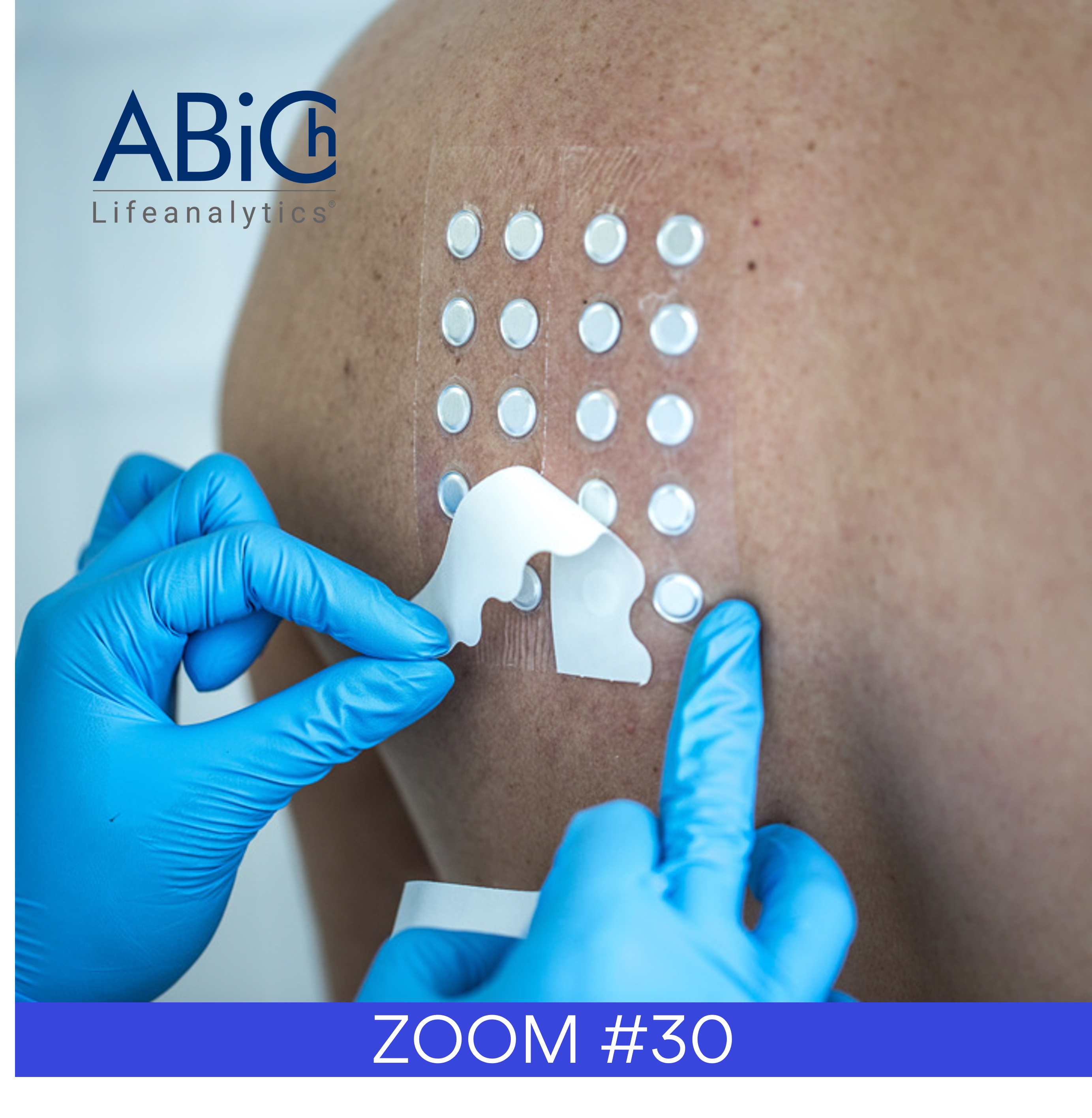
With our customers and/or specialized partners, we define the best model, ex-vivo, in-vitro, tissue engineering, skin model or subject sampling with or without microbiota and design the protocol to study the product activity. From clinical sampling, omics are useful for skin, hair or nails as anyway, it begins with the extraction of the necessary molecule. Then we produce the omics data from generated samples. The processes are optimized in order to generate the deepest investigation levels of omics, but screening approaches are also possible in order to get desirable cost constraints.
The huge amounts of data is then analysed with our bioinformatics pipelines. As the analytical technique is untargeted, the bioinformatic analysis is « hypothesis free »: Many effects are revealed including toxic effects, and possible claims will be deducted from these.
The only focusing of the tests is brought through the choice of the model on which the product will be applied.
CORAVALIDTM is the pipeline dedicated to human side only. It is a multi steps process which goes from an enrichment analysis up to and a final biological meanings and claims proposal.(6)
HolXploreTM is the pipeline dedicated to skin and its microbiome. It includes a taxonomic analysis of the diversity associated with different experimental conditions, up to functional analysis performed in parallel for bacteria, fungi and Homo sapiens taxons, correlations between different functions and/or taxons, and possible biological effects and claims.
Original claimings with original mechanisms can be so revealed.
Customized omics for cosmetics
For cosmetic science needs, our omics expertise is now diversified into more cosmetics focused offers.
Coupled with proteomics, phosphoproteomics is more and more an important indicator of the proteome activity. Protein phosphorylation is a crucial mechanism in many cellular regulations and strongly implicated in virtually all physiological and pathological processes
such as signal transduction, cell proliferation, differentiation, apoptosis or metabolism. Signaling processes involving phosphorylation are often deregulated during pathology or stress. The overall expression variations of the proteins are obtained and, for these, the variations of the phosphorylation motifs. This is a good indicator of a healthy skin.
To further understand the effects of abiotic stresses such as UV, blue light or pollution on the skin, RedOxmicsTM combines the proteomics approach to understand which pathways are involved in protection with a new dedicated data processing workflow, which can further provide an iterative approach of the most common 18 RedOx reaction modifications of 10 amino acids. This gives access to the OxDeepTM index which will indicate the stress deepness: The higher the ratio, more proteins are victims of oxidation.
As now the world discovered that our skin has a microbial part, PHYLOGENE dedicated specific offers to investigate this new domain :
- Follow-up by quantitative PCRs of major species or genus, depending on wished effects from product positioning. It gives answers such as: «The product has/does not have an effect on tested genus».
- Metagenomics comparative study of microbiome by NGSequencing 16S rDNA or ITS. It gives answers in a report as : «The product/treatment does or does not impact the semi-quantitative composition and microbiome diversity».
- Metaproteomics comparative study of microbiome by relative quantification LC-MS/MS « shotgun proteomics » and bioinformatics/biostatistics dedicated data processing and analysis for microbiome: HolXploreTM.
It gives answers in a report as : «Effects of the product on functions and interactions of host and microbiome simultaneously».
The omics and particularly those closest to the clinic are now essential to understand the mechanisms of action and obtain active ingredients that are efficient in cosmetics.
Bibliography
(1)Olsen JV and all. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 2013 Dec;12(12):3444-52
(2)Frankel AE and all. Metagenomic Shotgun Sequencing and Unbiased Metabolomic
Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune
Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia. 2017 Oct;19(10):848-855 (3)Hamzeiy H and all. What computational non-targeted mass spectrometry-based metabolomics can gain from shotgun proteomics. Curr Opin Biotechnol. 2017 Feb;43:141146
(4)Xiong W. and all. Development of an Enhanced Metaproteomic Approach for Deepening the Microbiome Characterization of the Human Infant Gut. J. Proteome Res. 2015; 14,
133−141
(5)Wang J and all. Proteome Profiling Outperforms Transcriptome Profiling for Coexpression
Based Gene Function Prediction. Mol Cell Proteomics. 2017 Jan; 16(1):121-134
(6)Hameury S and all. Prediction of skin anti-aging clinical benefits of an association of ingredients from marine and maritime origins: Ex vivo evaluation using a label-free quantitative proteomic and customized data processing approach. J Cosmet Dermatol. 2019
Feb;18(1):355-370
(7)Mills R and all. Evaluating Metagenomic Prediction of the Metaproteome in a 4.5-Year
Study of a Patient with Crohn’s Disease. mSystems 2019 Jan-Feb 4(1): e00337-18
(8)Starr A. and all. Proteomic and metaproteomic approaches to understand host-microbe interactions. Anal Chem. 2018 Jan 2;90(1):86-109.
web : https://www.phylogene.com
LinkedIn : https://www.linkedin.com/company/phylogene
Twitter : https://twitter.com/Phylogene_One





 Follow us on Linkedin!
Follow us on Linkedin!